Welcome to dbSNO 2.0!
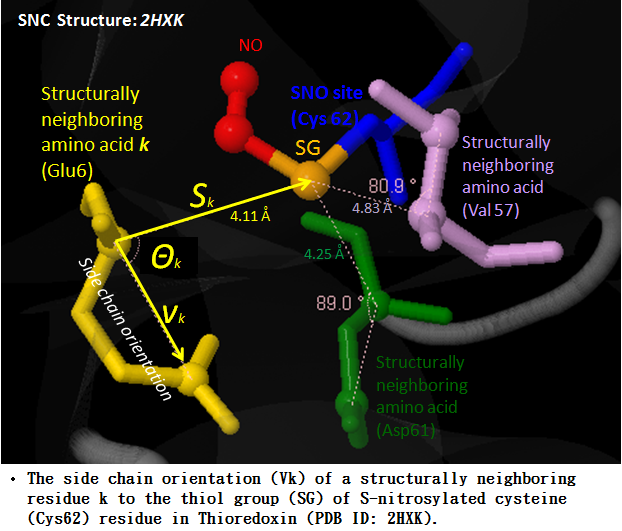
Protein S-nitrosylation (SNO) is a reversible post-translational modification (PTM) and involves the covalent attachment of nitric oxide (NO) to the thiol group of cysteine (Cys) residues. Given the increasing number of proteins reported to be regulated by this modification, S-nitrosylation is considered to act, in a manner analogous to phosphorylation, as a pleiotropic regulator that elicits dual effects to regulate diverse pathophysiological processes by altering protein function, stability, and conformation change in various cancers and human disorders. Due to its importance in regulating protein functions and cell signaling, dbSNO (http://dbSNO.mbc.nctu.edu.tw) is extended as an informative resource for exploring structural environment of SNO substrate sites and regulatory networks of S-nitrosylated proteins. An increasing interest in the structural environment of PTM substrate sites motivated us to map all manually curated SNO peptides (4165 SNO sites within 2277 proteins) to PDB protein entries by sequence identity, which provides the information of spatial amino acid composition, solvent-accessible surface area, spatially neighboring amino acids, and side chain orientation for 298 substrate cysteine residues. Additionally, the annotations of protein molecular functions, biological processes, functional domains and human diseases are integrated to explore the functional and disease associations for S-nitrosoproteome. In this update, users are allowed to search a group of interested proteins/genes and the system reconstructs the S-nitrosylation regulatory network based on the information of metabolic pathways and protein-protein interactions. Most importantly, an endogenous yet pathophysiological S-nitrosoproteomic dataset from colorectal cancer patients was adopted to demonstrate that dbSNO could discover potential SNO proteins involving in the regulation of NO signaling for cancer pathways.
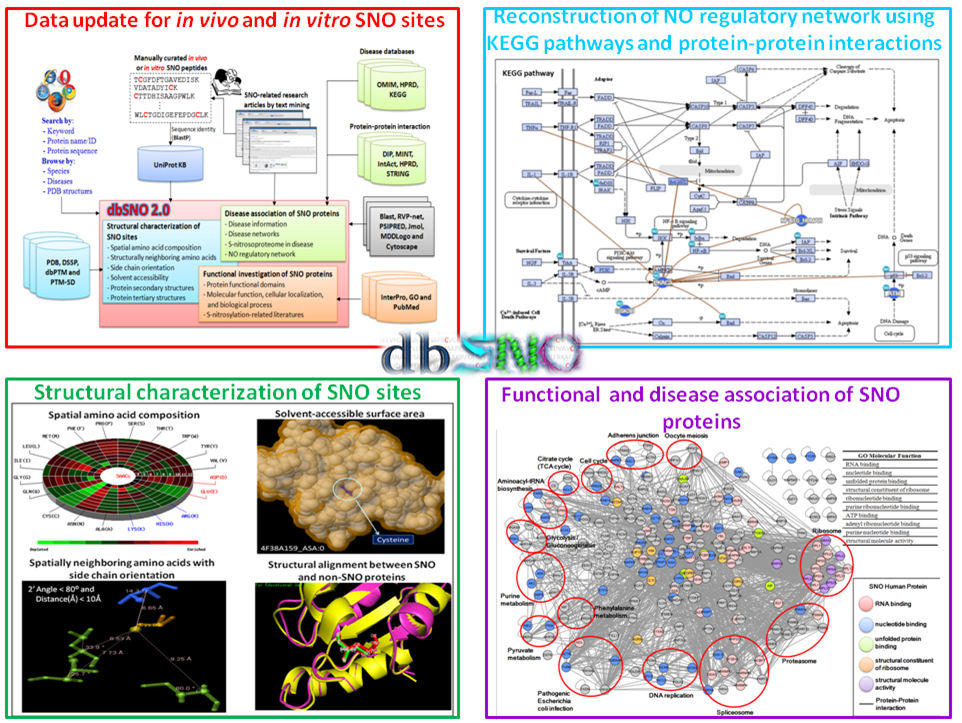
| The highlighted improvements and advances in dbSNO 2.0. | |
|
|
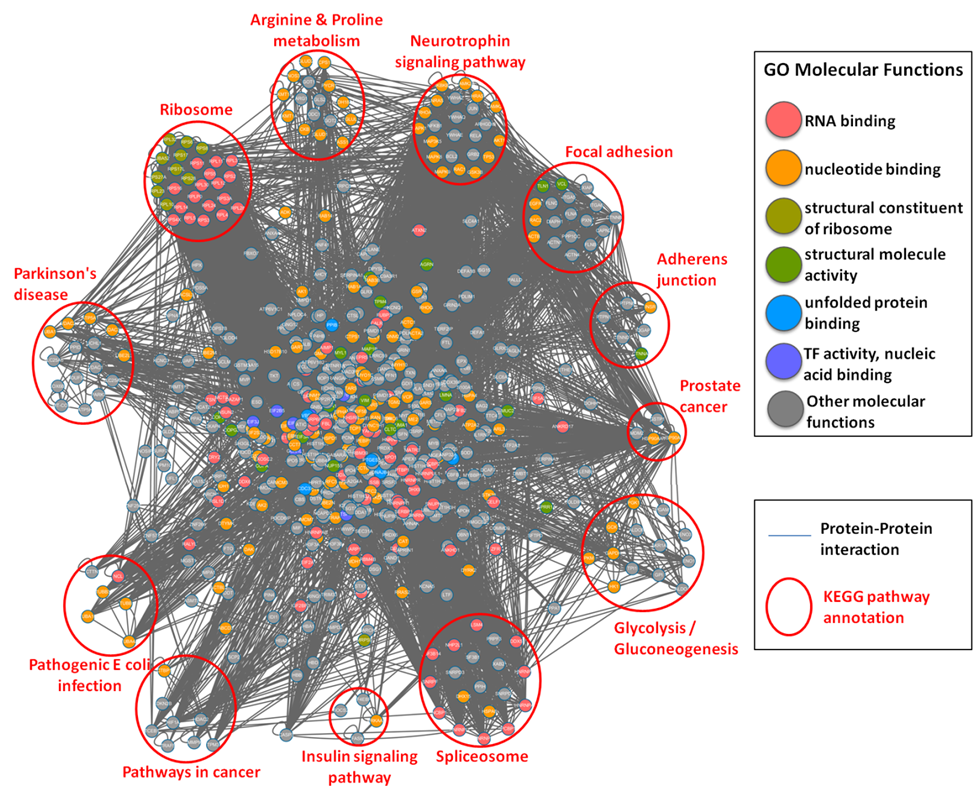
| The functional network of S-nitrosylated proteins in human. |
|
Protein-protein interaction network of all human S-nitrosylated proteins with the annotations of GO molecular function (marked in different colors) and KEGG metabolic pathways (clustered in different pathway groups). |
S-nitrosoproteomic Atlas in Individualized Human Colorectal Cancer Tissues
Yi-Ju Chen, Wei-Chieh Ching, Jinn-Shiun Chen, Tzong-Yi Lee, Cheng-Tsung Lu, Hsiao-Chiao Chou, Pei-Yi Lin, Kay-Hooi Khoo, Jenn-Han Chen, and Yu-Ju Chen (2014) "Decoding the S-nitrosoproteomic Atlas in Individualized Human Colorectal Cancer Tissues Using a Label-free Quantitation Strategy", Journal of Proteome Research, DOI: 10.1021/pr5002675. [PubMed]
| Workflow for site-specific identification and label-free quantitative approach for the S-nitrosoproteome |
|
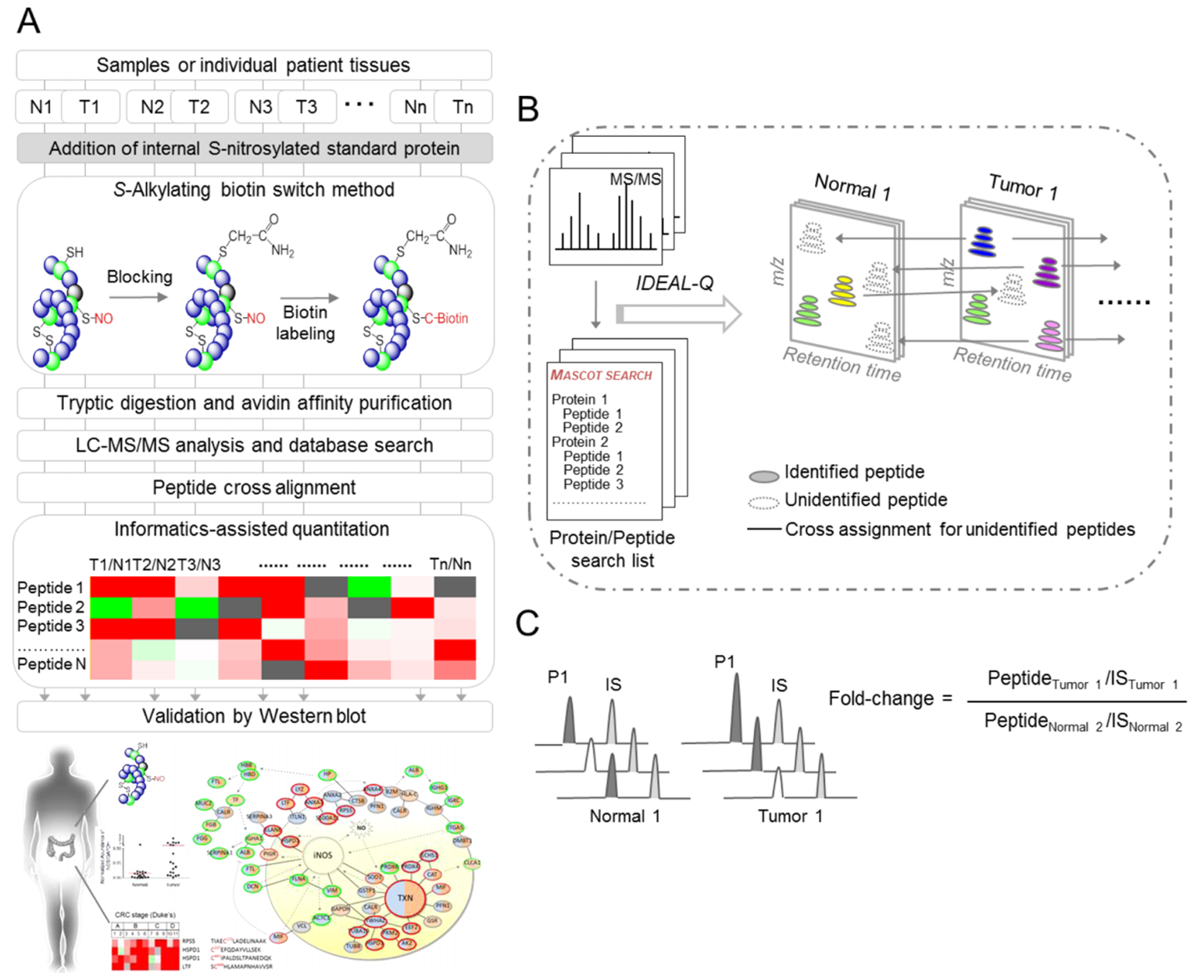
Total lysates of cells or paired tumorous and adjacent normal tissues from individual patients, were spiked with internal standard proteins that were S-nitrosylated in vitro. The S-nitrosylated proteins were modified by the S-alkylating biotin switch method and subjected to tryptic digestion, followed by streptavidin affinity purification. After triplicate LC-MS/MS analyses and data processing, the peptides were identified by Mascot database searches and peptide cross alignment, then further quantified by in-house IDEAL-Q software. The potential S-nitrosylated targets were further validated by western blot. N, normal tissue; T, tumor tissue. The total S-nitrosylated peptides were determined by the regressive alignment of the confidently assigned S-nitrosylated peptides based on the same elution time and cross-assignment for the same S-nitrosylated peptides in all LC-MS/MS runs. To calculate the S-nitrosylated peptide ratio, the peptide abundance was first normalized to the XIC peak area of the internal S-nitrosylated standard peptide (IS) in the same LC-MS/MS run. The fold change in each peptide was calculated by the normalized peptide abundance in different samples. |
Site-speci?c Identi?cation of the S-nitrosoproteome
Yi-Ju Chen, Wei-Chi Ku, Pei-Yi Lin, Hsiao-Chiao Chou, Kay-Hooi Khoo, and Yu-Ju Chen* (2010) "S-Alkylating Labeling Strategy for Site-Speci?c Identi?cation of the S-Nitrosoproteome", Journal of Proteome Research, Vol. 9(12):6417-39. [PubMed]
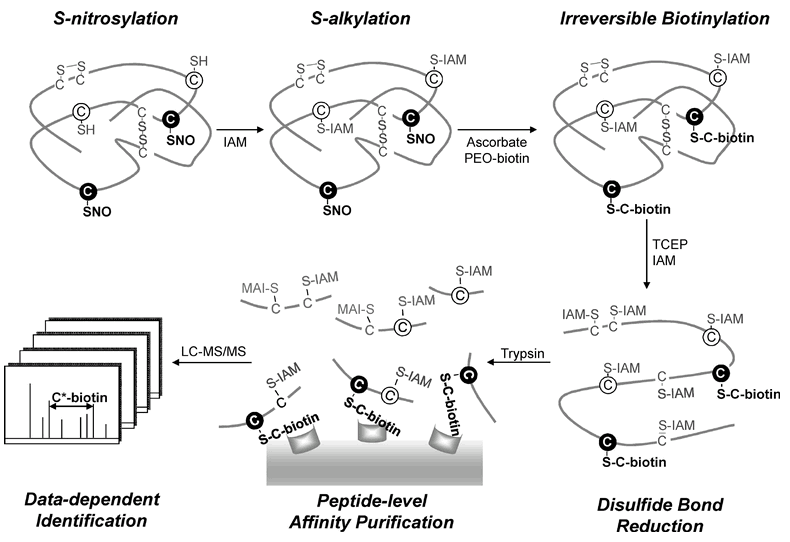
| Workflow for the site-speci?c identi?cation of the S-nitrosoproteome by a modi?ed biotin switch method |
|
Three forms of cysteine residues on an S-nitrosylated protein are shown: free thiols (open circles), disul?de bonds (solid lines), and S-nitrosylated thiols (?lled circles). The free cysteine thiols were ?rst blocked by S-alkylation with IAM. Site-speci?c labeling of the S-nitrosylated cysteine thiols was performed by reduction with ascorbate followed by irreversible biotinylation with PEO-iodoacetyl-biotin. The disul?de bonds were further reduced by TCEP and S-alkylated by IAM. After tryptic digestion, the biotinylated peptides were enriched by avidin af?nity puri?cation and analyzed by LC-MS/MS. Based on the characteristic mass shift (517.2 Da) of the fragment ion from a biotinylated cysteine residue in the MS/MS spectrum, the S-nitrosylation site could be unambiguously determined. |